Analyse de l’eau
1) pH et alcalinité
Principe du dosage
Il s’agit d’un titrage d’une base faible par un acide fort.
On effectue un titrage pHmétrique de l’eau à analyser par une solution d’HCl 20 mmol.L-1 et on détermine le point d’équivalence.
Le point d’équivalence peut être déterminé par le virage d’indicateurs colorés (Tableau 1. Méthylorange ou vert de bromocrésol) ou par la réalisation d’une courbe de titrage (Fig.1).

Théoriquement, le point d’équivalence correspond au point d’inflexion de la courbe de titrage. Mathématiquement, pour une fonction décroissante comme c’est le cas ici, il s’agit du minimum de la dérivée première de la courbe (voir Fig. 9), ce qui correspond au minimum de la pente de la tangente à un point de cette courbe.
Sur la Fig. 1, le point d’équivalence se situe au pH 4,41 pour un volume de titrant de 15,93 mL (le point de la courbe de titrage où la pente (m) est minimale : -2,22).
En pratique, ce point n’est pas toujours exactement situé à un pH de 4,3 (ici 4,32) mais cela n’a toutefois pas une grande incidence au niveau de la différence du volume de titrant (ici 0,03 mL) et des calculs qui s’en suivent.
Première méthode (visuelle) : indicateurs colorés
Montage: voir Fig. 2.
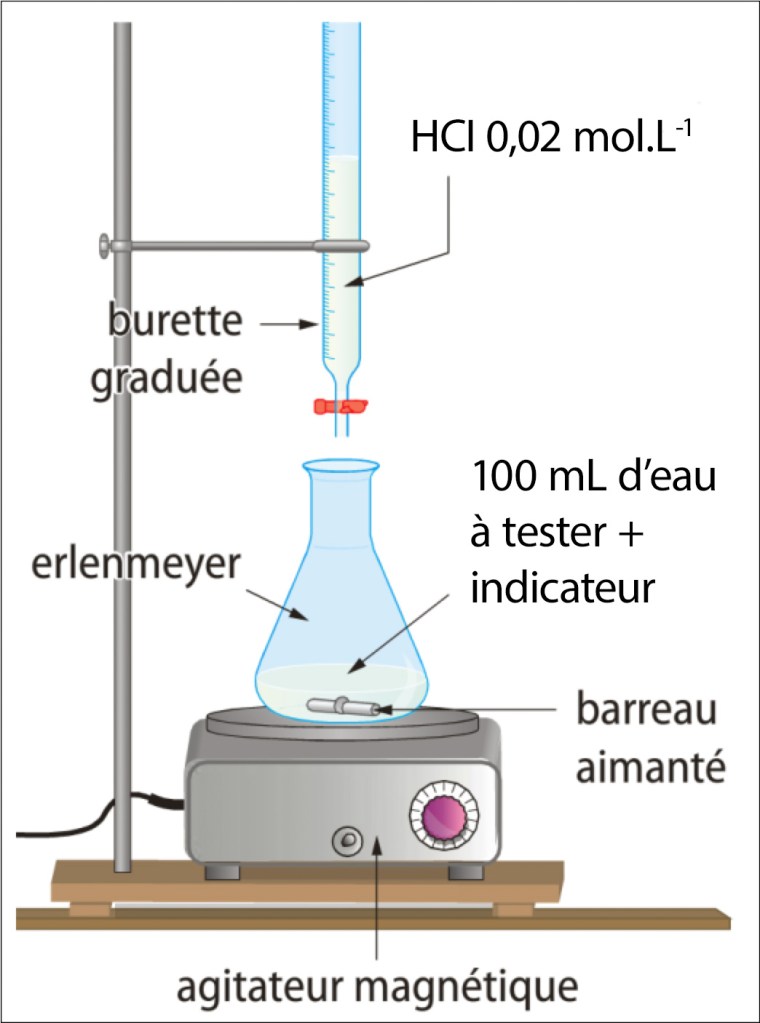
- Dans un bécher (erlenmeyer ou tout autre récipient) de 250 mL, verser un volume V0 = 100 mL d’eau à tester (échantillon)
- Rajouter un turbulent (barreau aimanté) et environ 20 gouttes de phénolphtaléine (sol. méthanolique 1%). Observer la coloration prise par la solution.
- Si la solution n’est pas colorée, rajouter environ 20 gouttes de vert de bromocrésol (sol. aqueuse 0,04%) ou de méthyl orange (sol. aqueuse 0,04%) dans le bécher.
- Après l’avoir rincée, remplir une burette de 50 mL avec la solution d’acide chlorhydrique (20 mmol.L–1) et faire le zéro.
- Placer le bécher sur un agitateur magnétique et le tout sous la burette graduée.
- Verser l’acide chlorhydrique goutte à goutte jusqu’à atteindre la teinte sensible du vert de bromocrésol (ou au méthyl orange) : le changement de couleur doit être persistant et se faire à la goutte près.
- Noter le volume d’acide versé lorsque l’indicateur coloré a changé de couleur : on est à l’équivalence.
Remarque
Il faut bien tenir compte des zones de virages des indicateurs :
| Indicateur coloré | Teinte acide | pH zone de virage (teinte sensible) | Teinte basique |
|---|---|---|---|
| Méthyl Orange (MO) | Rouge | 3,1 Orange 4,4 | Jaune |
| Phénolphtaléine | Incolore | 8,2 Rose 9,9 | Violet |
| Vert de Bromocrésol (VB) | Jaune | 3,6 Vert 5,4 ≈ 4,55 | Bleu |
Les solutions étant fort diluées, les teintes sont assez pâles et il convient de cesser le titrage lorsque la couleur de l’échantillon à doser vire à l’orange (MO) ou au jaune (VB) persistants. On obtient à peu près les mêmes volumes. Dans le cas du VB, il est préférable de choisir comme pH équivalent une valeur intermédiaire (4,55), plus proche de la réalité.
On obtient des valeurs plus précises en réalisant un courbe de titrage pHmétrique (voir ci-dessous).
Deuxième méthode : pHmétrie

- Le montage est identique à celui mis en place pour les indicateurs colorés excepté que l’on introduit une électrode de pH dans le bécher contenant les 100 mL d’eau à tester (voir Fig. 3.).
- Dans cette méthode, au lieu de ne tenir compte que du volume d’acide ajouté jusqu’au changement de couleur de l’indicateur, on réalise ce qu’on appelle une courbe de titrage.
- Cette courbe peut se construire de deux manières : manuelle ou automatique.
- Manuelle : on ajoute le titrant (HCl 20 mmol.L-1) millilitre par millilitre en reportant sur un graphique le pH en fonction du volume d’HCl ajouté. On ralentit le débit de la burette lorsqu’on s’approche de pH 5 (le point d’équivalence se situe vers pH 4). On poursuit le titrage jusque pH 3 environ.
- Automatique : on relie la sonde de pH à une interface transformant un signal analogique en un signal digital. Cette interface est connectée à un ordinateur muni d’un logiciel d’acquisition de données qui affiche en temps réel la variation de pH en fonction du temps (et non du volume !)2. Dans ce cas, il est impératif de titrer selon un débit de la burette régulier et continu (goutte à goutte – ne pas manipuler le robinet pendant le titrage !) ; l’agitation doit être assez intense afin de bien homogénéiser la solution titrée. Il faut aussi noter visuellement le volume initial (Vi) et final (Vf) du titrage.
Traitement des données
Les données des titrages sont acquises via l’application SPARKvue® v4.53, dédiée à l’utilisation du matériel didactique PASCO4. Ce logiciel permet de paramétrer les expériences, de traiter les données acquises et d’exporter les celles-ci, qui peuvent éventuellement être étudiées plus en détail sous tableur (Microsoft® Excel, Apple® Numbers, Google Sheets …)5.
Le système SPARKVue® exporte les données d’acquisition alphanumériques sous un format .csv. Pour être exploitables, elles doivent être converties dans le format ad hoc (Excel .xslm par ex.).
Le capteur « Advanced Chemistry Sensor » enregistre, en plus des paramètres nécessaires (ici le temps et le pH), plusieurs autres données non applicables. Dans un souci de simplification de la feuille de calcul, il convient de supprimer d’emblée les colonnes non utilisées.
Deux options de titrage sont possibles : avec ou sans compte-goutte connecté.
Première option : mesures sans compte-goutte
Opération préalable : transformation du temps (secondes) en unités de volume (mL).
On note Vi le volume affiché sur la burette au début du titrage au temps initial Ti et Vf le volume indiqué en fin de titrage au temps final Tf. Une seconde correspond alors à :
1 sec ⇒
Voici une capture d’écran représentant en temps réel la courbe du titrage de 100 mL d’eau d’Évian par HCl 20 mmole.L-1 :

La fréquence d’acquisition est de 1 Hz soit une mesure de pH par seconde.
Dans le cas présent :
Vi = 0,1 mL et Vf = 32,3 mL.
Ti = 0 sec et Tf = 302 sec.
1 sec correspond donc à :
32,2/302 = 0,10662 mL.
Deuxième option : utilisation d’un compte-goutte
Opération préalable : calibrage du compte-goutte. Pour obtenir une courbe de titrage représentant le pH directement en fonction du volume de titrant ajouté, il faut déterminer précisément le volume des gouttes que le dispositif compte6. Cette manipulation, bien que nécessaire peut parfois s’avérer chronophage…
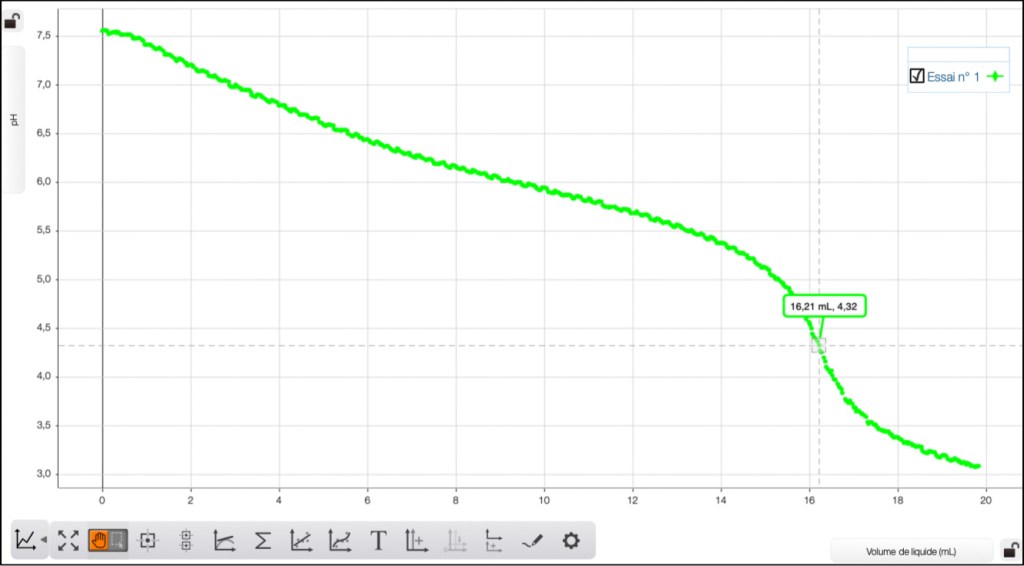
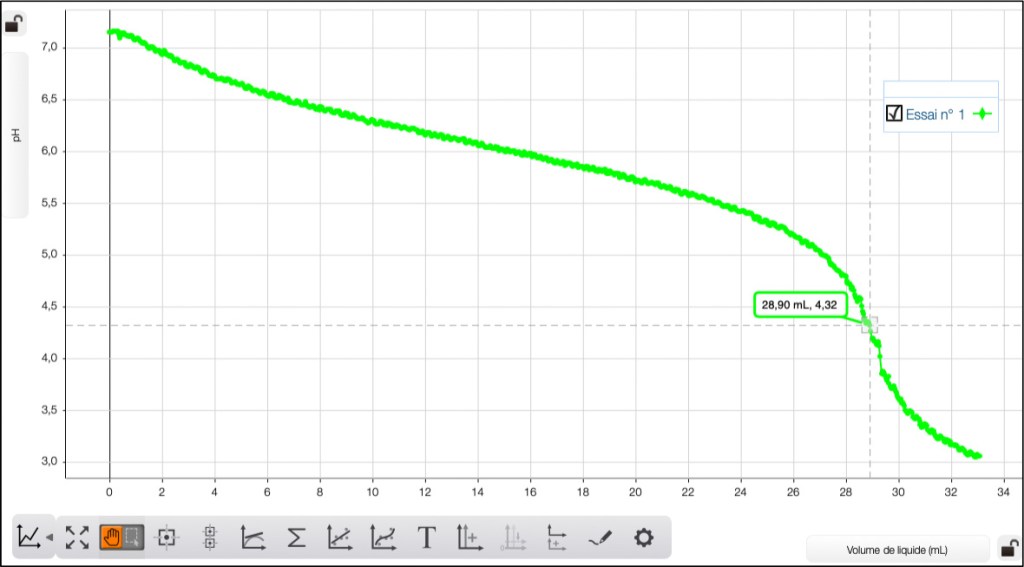
Voici les courbes enregistrées relatives aux 4 eaux étudiées (Fig. 5-8):
Eau de distribution (robinet)
Eau de boisson (Evian)
Eau de fleuve (Muga)

Eau de mer
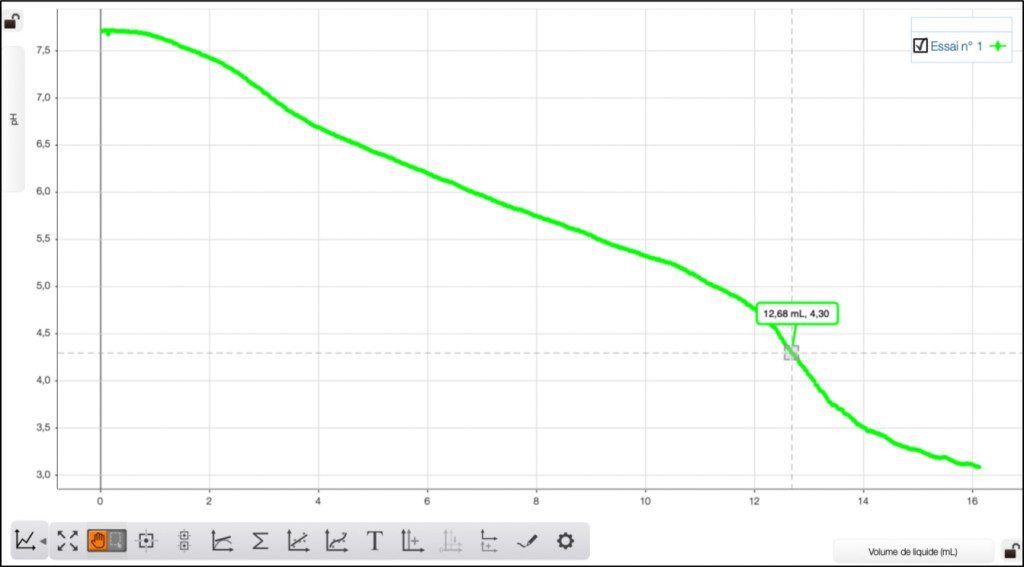
Pour être conforme à la définition de la KS4,3 , on note le volume équivalent à pH 4,3 soit pour :
- l’eau de distribution : 16,21 mL
- l’eau d’Évian : 28,9 mL
- l’eau de fleuve : 21,78 mL
- l’eau de mer : 12,68 mL
Détermination précise du point d’équivalence
Dans Excel, il faut insérer une nouvelle colonne à côté du temps et y introduire les valeurs correspondantes des volumes de titrant ajoutés. Il est maintenant possible de créer un graphique représentant une courbe titrage : pH = f(V)
Le point d’équivalence (Véq ; pHéq) se trouve au point d’inflexion de cette courbe. Pour déterminer exactement les coordonnées de ce point, on utilise la méthode des dérivées.
La fonction dérivée se construit de la manière suivante : pour chaque point i de la courbe de titrage, on calcule (tableur) :

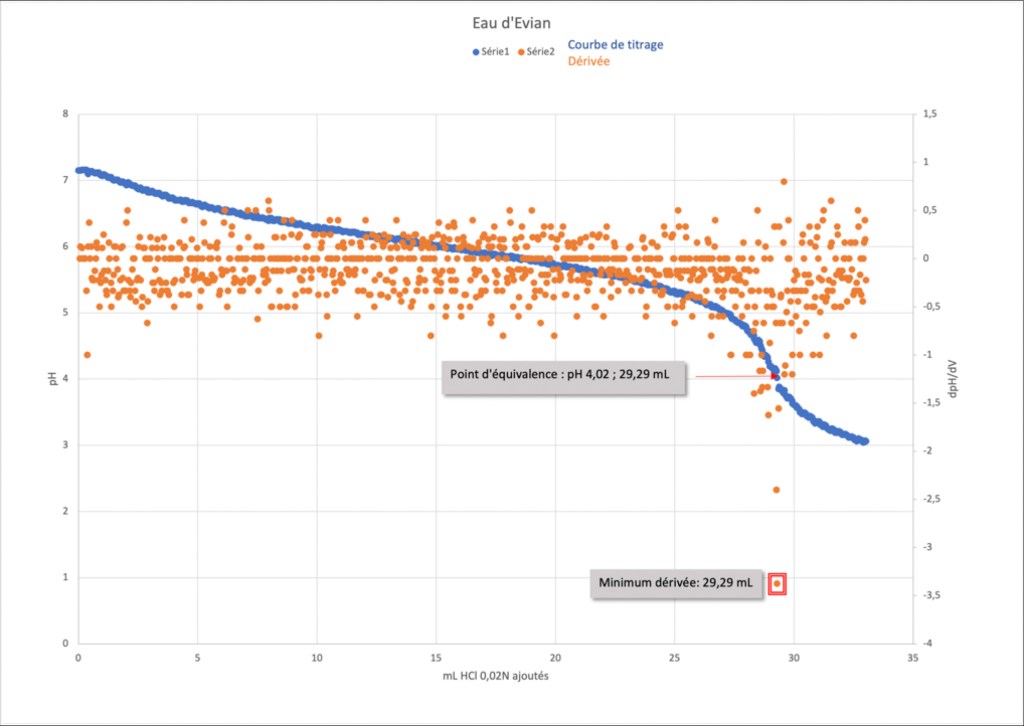
On obtient un nuage de points (courbe dérivée) et la valeur du volume Véq et du pHéq se trouve au minimum de celle-ci (cas du titrage d’une base faible par un acide fort). Pour l’eau d’Evian, le point d’équivalence a donc pour coordonnées (29,9 ; 4,02, voir graphique ci-contre7).
Remarque
En général, le point d’inflexion ne se situe pas exactement à pH 4,3 (ici pH 4,02).
Cela n’a toutefois pas une grande incidence au niveau du volume de titrant (ici 0,39 mL, soit 1,35 %) et permet d’obtenir pratiquement les mêmes résultats.
2) La dureté
Le principe est celui d’un titrage complexométrique. Le montage est celui d’un titrage direct (voir figure 10.).
Principe du dosage
La mesure du titre hydrotimétrique (TH) est basée sur un titrage complexométrique8. Le dosage s’effectue avec une solution de sel di-sodique de l’acide éthylène diamine tétraacétique (Na2EDTA) symbolisé par Na2H2Y. L’anion H2Y2-est un ion complexe qui donne avec de nombreux cations, des composés stables mais incolores. Aussi doit-on utiliser un indicateur de fin de réaction pour mettre en évidence la fin du dosage par le Na2EDTA : le noir d’ériochrome T (NET). Cet indicateur sera utilisé pour doser les ions Ca2+ ou Mg2+.
Les réactions de complexation s’écrivent :
Ca2+ + Na2H2Y ⇋ [CaH2Y] + 2Na+
Mg2+ + Na2H2Y ⇋ [MgH2Y] + 2Na+
Pour que ces réactions puissent être utilisées pour le dosage de ces ions (on les dose ensemble), il faut procéder à un pH voisin de 10, où la forme Na2H2Y est majoritaire.
Le virage du NET se fait du rose violacé (présence de Ca2+ et de Mg2+) au bleu : Ca2+ et de Mg2+ étant totalement complexés par le Na2EDTA.
Protocole expérimental
Préparation des solutions :
Solution titrante : Na2EDTA 0,01 mol.L-1 9
Tampon ammoniacal (500 mL) : Mélanger 250 mL d’une solution aqueuse d’ammoniac (NH3)10 à 1 mol.L-1 et 250 mL d’une solution de chlorure d’ammonium (NH4Cl) 1 mol.L-1
Indicateur : NET à 1 % dans l’alcool éthylique 5-10 %
Solution à titrer : 100 mL d’eau à analyser (échantillon)
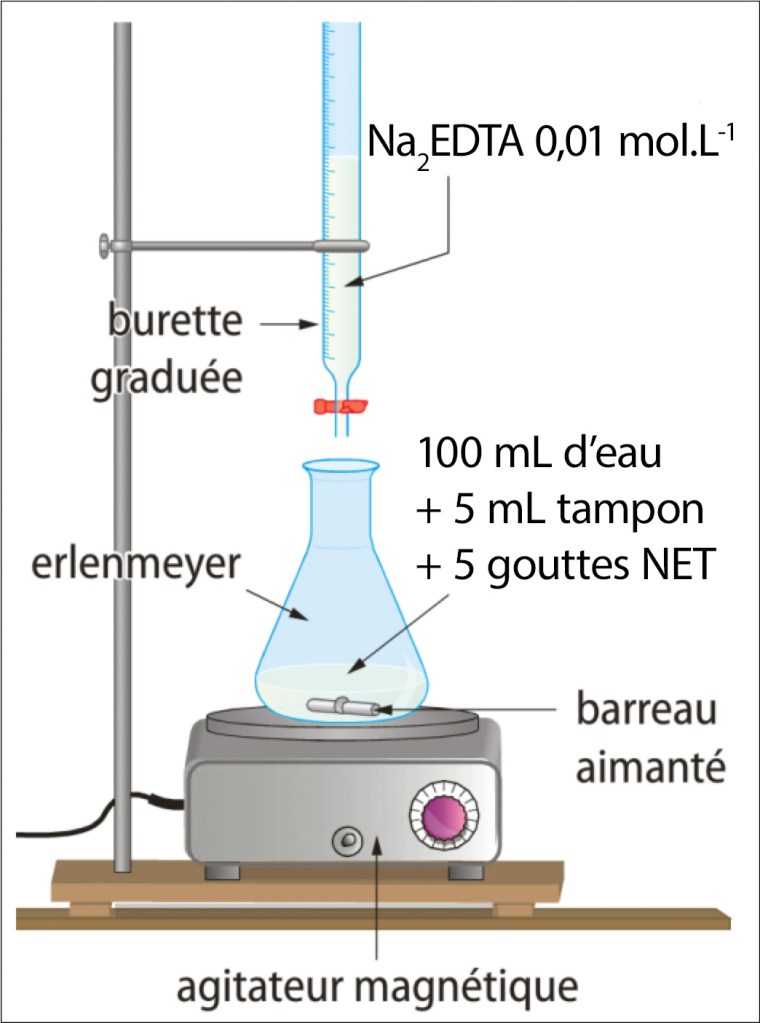
- Après avoir rincé la burette avec la solution de Na2EDTA, la remplir jusqu’au trait et ajuster le zéro.
- Remplir un bécher de 250 mL (ou tout autre récipient) avec l’échantillon et chauffer celui-ci à 40°C. La plupart des agitateurs magnétiques sont munis d’une plaque chauffante. Contrôler la température avec un thermomètre.
- Ajouter 5 mL de solution tampon et 5 gouttes de NET.
- Déposer le bécher sur un agitateur magnétique et y introduire un barreau aimanté.
- Commencer le titrage jusqu’au virage du rose violacé au bleu. Arrêter l’addition de Na2EDTA et noter le volume. La première manipulation est souvent peu précise.
- Effectuer un second dosage précis ; l’ajout de titrant devant se faire goutte à goutte au niveau de la zone de virage. Noter le volume et utiliser cette valeur pour effectuer les calculs.
Calcul de la dureté totale
Soit :
CE la concentration en Na2EDTA = 0,01 mol.L-1
VE le volume de Na2EDTA consommé lors du titrage (en mL)
Veau le volume d’échantillon d’eau = 100 mL
Ceau la somme des concentrations en Ca++ et Mg++ en mol.L-1 dans l’échantillon
On a : CE × VE = Veau × Ceau , d’où

d’où Ceau = 10-4 .VE mol.L-1 = 10-1 .VE mmol.L-1.
Dans ce cas précis, VE donne directement le TH °f (sans unité).
3) Dosage des sulfates
Principe du dosage
Principe de base :
Les anions SO42- réagissent avec les cations baryum Ba2+ en formant un précipité blanc de sulfate de baryum BaSO4 insoluble (pKs = 9,96) et quantifiable.
En pratique, on fait réagir une solution de chlorure de baryum BaCl2 de concentration connue avec la solution d’ions SO42- de concentration inconnue.
BaCl2(aq) + SO42-(aq) → BaSO4(s) + 2Cl–(aq)
Dès que la solution de BaCl2 entre en contact avec la solution contenant des ions sulfate, un précipité blanc de sulfate de baryum (BaSO4) apparait. L’équivalence est obtenue quand n(Ba2+) = n(SO42-). Comme le précipité se forme dès le début du mélange des deux solutions, et qu’il n’existe pas d’indicateur coloré indiquant un excès d’ion Ba2+, il est nécessaire d’utiliser une autre technique. Deux méthodes sont couramment utilisées basées sur ce principe : la turbidimétrie et le titrage conductimétrique.
1- Méthode par turbidimétrie
La turbidimétrie est la mesure du degré de turbidité d’une suspension. Elle peut être déterminée à l’aide d’un spectrophotomètre classique qui mesure la diminution, due à l’absorbance, de l’intensité d’un rayon lumineux de longueur d’onde connue traversant la suspension11. Si le volume des particules est maintenu constant, on peut appliquer une loi identique à la loi de Lambert-Beer :

avec :
A : absorbance de la suspension (grandeur sans dimension)
Io : intensité lumineuse du faisceau incident
I : intensité lumineuse du faisceau transmis
ε : constante propre à la suspension
l : trajet optique (en général, on utilise des cuvettes de 1 cm d’épaisseur)
C : la « concentration » en particules en suspension, c-à-d la turbidité
On voit que l’absorbance est directement proportionnelle à la turbidité, du moins pour les suspensions peu concentrées.
Remarque
Il faut s’assurer que la suspension reste homogène pendant les mesures. Pour cela il faut garder la taille des particules de BaSO4 constante et éviter leur décantation. Un mélange de glycérine et d’alcool et de sels solubles (force ionique élevée) sert à stabiliser les particules et à empêcher une sédimentation rapide de celles-ci.
2- Méthode par conductimétrie
Il s’agit d’un dosage volumétrique par précipitation. On réalise un titrage conductimétrique : on utilise une cellule de conductimétrie qui mesure en temps réel l’évolution de la conductivité de la solution titrée (contenant les ions SO42- à doser) au fur et à mesure de l’ajout de BaCl2 et on enregistre les variations de conductivité en fonction du volume de titrant ajouté.
Cette méthode ne sera pas utilisée ici car les résultats sont souvent difficiles à interpréter.12
Le dosage se fera donc par turbidimétrie en utilisant une droite d’étalonnage à 6 points.
Le tableau suivant reprend en détail les différentes étapes de la préparation des milieux réactionnels aboutissant à l’établissement de la droite permettant d’évaluer la concentration en sulfates de la solution inconnue.
| Étape 1 : préparation de solution étalon en sulfate | ||||
| MM (g.mol-1) | Sol stock : 10-3 mole.L-1 SO42- 1 mg.l-1 = 10-5 mol.L-1 Sol stock: 100 mg.L-1 SO42- | |||
| MgSO4.7H2O | 246,47 | 0,246 g.L-1 à préparer | ||
| [SO4—] | 96,06 | 96,06 mg.L-1 | ||
| Étape 2 : préparation du réactif | Remarque | |||
| Pour 500 mL de réactif, mélanger : | Si ces substances contiennent des traces de sulfates, un léger trouble peut déjà apparaître lors de la préparation du réactif. Si c’est le cas, laisser reposer celui-ci au moins 24 h avant de faire les mesures. Ne pas agiter ! | C’est souvent le cas lors de l’utilisation de produits « ménagers ». | ||
| H2O | 300 mL | |||
| Ethanol 96% | 100 mL | |||
| Glycerol | 50 mL | |||
| HCl (6 M) | 30 mL | |||
| KCI ou NaCl | 70 g | |||
| BaCl2 | 20 g | |||
| Étape 3 : préparation des milieux réactionnels | ||||
| Dans béchers (ou autres récipients de format identique) de 100 mL introduire : | ||||
| N° récipients | Eau distillée (mL) | Sol. stock (mL) | Réactif (mL) | [SO4—] (mg.L-1) |
| 1 | 50 | 0 | 5 | 0 |
| 2 | 45 | 5 | 5 | 10 |
| 3 | 40 | 10 | 5 | 20 |
| 4 | 35 | 15 | 5 | 30 |
| 5 | 30 | 20 | 5 | 40 |
| 6 | 25 | 25 | 5 | 50 |
| Selon le nombre d’échantillons de concentration inconnue | ||||
| 50mL | 5 | ? | ||
| Étape 4 : après homogénéisation des milieux, lecture au spectrophotomètre à 567 nm (cuvettes de 1 cm) | ||||
NB : tout autre sulfate soluble peut être utilisé pour l’étalonnage (Na2SO4 ou K2SO4 par ex.).
Remarque
Les valeurs d’absorbance obtenues dépendent fortement de la préparation des solutions.
La mesure de l’absorbance peut se faire à n’importe quelle longueur d’onde (ici 567 nm). En effet, on ne mesure pas une absorbance due à une solution colorée (transition électronique), mais à une perte d’intensité lumineuse par turbidité.
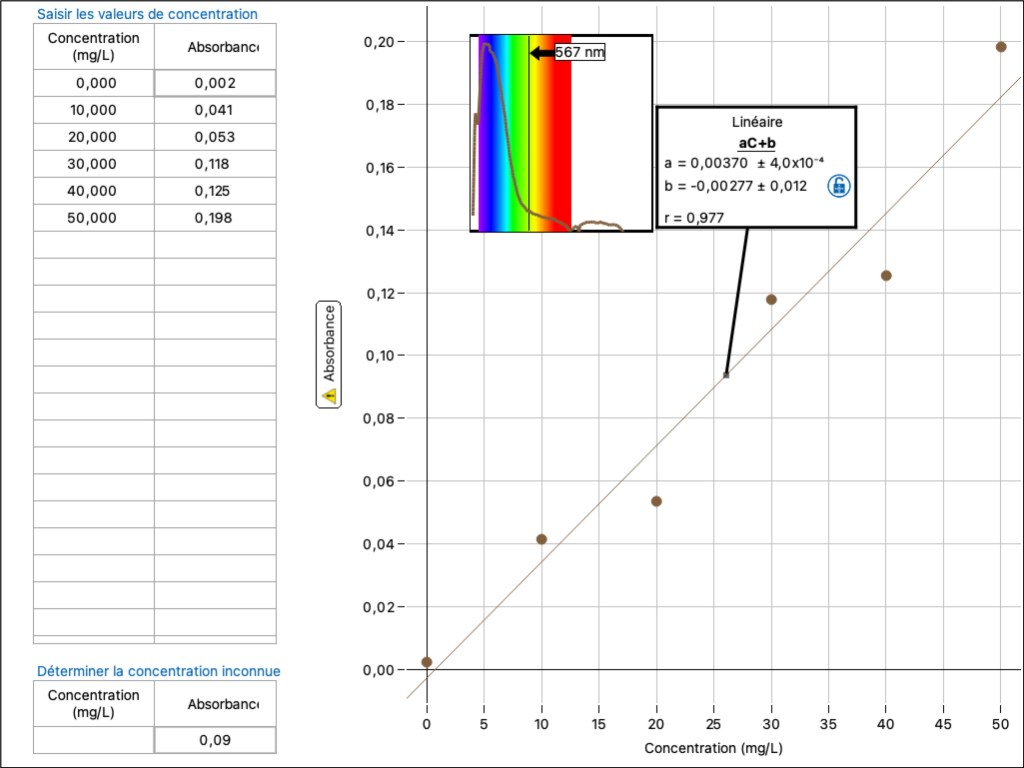
Les mesures sont obtenues avec un spectrophotomètre PASCO13. Les données sont acquises et analysées à l’aide du logiciel « Spectrometry »14. Voici l’allure de la droite d’étalonnage.
L’ajustement linéaire donne l’équation de celle-ci (A = aC + b) et son coefficient de corrélation.
Dans le cas présenté ci-contre, la concentration inconnue a une absorbance de 0,09, ce qui correspond à une valeur de 24,3 mg.l-1 (eau d’Evian).
4) Dosage des nitrates
Principe du dosage15
En milieu anhydre, l’action de l’acide sulfurique sur le salicylate de sodium donne avec les nitrates un mélange d’ortho- et de para-nitrosalicylate de sodium. En milieu basique, l’anion nitrosalicylate est libéré et sa coloration jaune stable permet un dosage colorimétrique à la longueur d’onde de 415 nm.
L’adjonction de tartrate double de sodium et potassium en même temps que la soude empêche la précipitation des sels de calcium et magnésium.
Cette méthode de dosage colorimétrique des ions nitrates est applicable pour des concentrations en ions NO3– comprises entre 0,15 et 15 mg.L-1.
Réactions : le mécanisme de celles-ci est typiquement une substitution électrophile aromatique (nitration)16 dont voici le bilan simplifié :
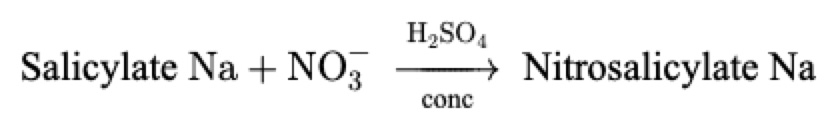
Le dosage se fait par spectrophotométrie (matériel PASCO) en utilisant une droite d’étalonnage à 6 points.
Le tableau suivant reprend en détail les différentes étapes de la préparation des milieux réactionnels aboutissant à l’établissement de la droite permettant d’évaluer la concentration en nitrates de la solution inconnue.
| Étape 1 : Préparation de la solution étalon en nitrate | ||||
| MM g.mol-1 | Sol. stock : 100 mg.L-1 NO3– | |||
| KNO3 | 101,1 | 163 mg.L-1 à préparer | ||
| NO3– | 62 | 100 mg.L-1 | ||
| Étape 2 : préparation des milieux réactionnels phase 1 | ||||
| Dans béchers (ou autres récipients identiques en verre) de 100 mL introduire : | ||||
| N° récipients | Eau distillée (mL) | Sol. stock (mL) | [NO3–] mg.L-1 | Na salicylate 0,5 % (mL) |
| 1 | 10 | 0 | 0 | 1 |
| 2 | 9,5 | 0,5 | 5 | 1 |
| 3 | 9 | 1 | 10 | 1 |
| 4 | 8,5 | 1,5 | 15 | 1 |
| 5 | 8 | 2 | 20 | 1 |
| 6 | 7,5 | 2,5 | 25 | 1 |
| 7 … Échantillons de concentration inconnue | 10 mL | ? | 1 | |
| Étape 3 : Faire évaporer à sec ces solutions dans un four à microondes programmé à faible puissance pour éviter l’ébullition! | ||||
| Étape 4 : préparation des milieux réactionnels phase 2 | ||||
| Après évaporation complète, reprendre le résidu refroidi en ajoutant dans chaque récipient, dans l’ordre : | ||||
| 1 ⇒ | 2 ⇒ | 3 | Étape 5 | |
| H2SO4 98% (mL) | H2O dist (mL) | Réactif de coloration (mL) | Lecture au spectrophotomètre à 425 nm après homogénéisation et refroidissement (cuvettes de 1 cm). | |
| 2 | 15 | 15 | ||
| Laisser réagir 10 min en agitant délicatement | Ajouter très progressivement | Ajouter très progressivement | ||
| Attention ! Risques de projections dues à l’augmentation brutale de température | ||||
| Réactif de coloration : | ||||
| 40 % NaOH et 6 % tartrate double de Na & K | ||||
| en solution aqueuse (eau distillée). | ||||
5) La salinité de l’eau de mer
Principe des dosages
1. Volumétrie par titration
Cette méthode est basée sur la loi de Dittmar. On dose la teneur en halogénures (principalement les chlorures et les bromures) que l’on exprime en masse (g) de chlore équivalente à la masse totale des halogènes présents dans 1 kg d’EdM ; cette valeur est appelée chlorinité. Celle-ci est multipliée par un facteur de 1,80655 qui représente le rapport de la salinité moyenne (S = 35) sur la quantité de chlore présente dans l’EdM qui est de 19,37394 g.kg-117.
Principe de la méthode de Mohr18 :
Il s’agit d’un dosage argentimétrique par précipitation. On a une solution d’ions chlorures (halogénures) de concentration inconnue. On ajoute dans cette solution du nitrate d’argent AgNO3. A chaque quantité d’ion argent (Ag+) ajouté, se forme un précipité de chlorure d’argent (AgCl) :
Ag+(l) + Cl–(l) → AgCl(s)↓
La solution transparente au départ se trouble d’une opalescence blanche pour devenir ensuite opaque.
L’équivalence est atteinte à l’équilibre stœchiométrique : n(Ag+) = n(Cl–). Comme rien ne permet de visualiser cette équivalence, le trouble blanc présent depuis le début du titrage ne changeant plus, on utilise un indicateur coloré de la présence d’ions Ag+ en excès. On utilise du chromate de potassium (K2CrO4) en solution aqueuse qui forme, dès que les ions Ag+ sont en excès, un précipité rouge brique de chromate d’argent (Ag2CrO4).
Remarque importante :
Certains sels d’argent sont fort sensibles à la lumière19 ; il faut conserver leurs solutions dans des flacons en verre brun ou emballés dans du papier aluminium, sinon on observera progressivement un noircissement de celles-ci. En outre, le contact d’AgNO3 avec la peau provoque des taches noires indélébiles. Il est fort conseillé de manipuler le nitrate d’argent avec des gants et des lunettes de protection !
Mode opératoire
Le montage est celui d’un titrage direct.
Préparation des solutions :
Solution titrante :
Solution aqueuse de AgNO3 5.10-2 mol.L-1 .21
Indicateur :
Solution aqueuse de K2CrO4 à 5%.
Solution à titrer :
Échantillon d’EdM non dilué.
1. Après avoir rincé la burette avec la solution d’AgNO3, la remplir jusqu’au trait et ajuster le zéro.
2. Dans un bécher de 250 mL (ou tout autre récipient permettant une titration) on introduit 1 mL d’échantillon d’EdM non dilué et on ajoute environ 20 mL d’eau distillée20 et on ajoute finalement 5 mL de la solution de K2CrO4 .
3. Déposer le bécher sur un agitateur magnétique et y introduire un barreau aimanté.
4. Commencer le titrage jusqu’au virage du jaune au rouge. Arrêter l’addition de AgNO3 et noter le volume équivalent VE. La première manipulation est souvent peu précise. Effectuer un second dosage plus précis ; l’ajout de titrant devant se faire goutte à goutte au niveau de la zone de virage.
Remarque : la zone de virage ne se fait pas instantanément mais passe par une couleur orange qui apparait au voisinage du point d’équivalence.
2. Titrage conductimétrique
Dans ce cas, on n’utilise pas d’indicateur de précipitation coloré mais une cellule de conductimétrie qui mesure en temps réel l’évolution de la conductivité de la solution titrée au fur et à mesure de l’ajout d’AgNO3. Cette opération peut s’effectuer sans calibration préalable de la sonde car on observe des variations de conductivité et non des valeurs absolues.
Méthode – Système et didacticiel PASCO:
1. Dans un bécher de 250 mL on introduit 1 mL d’échantillon d’EdM sur lequel on ajoute 175 mL d’eau distillée.
2. Introduire la sonde de conductimétrie dans le bécher de manière à ce que la cellule de mesure soit complètement immergée. Éliminer soigneusement toute présence de bulles d’air pouvant faire obstacle à la circulation des ions et démarrer l’agitation (barreau aimanté ou dispositif solidaire de la cellule).
3. Laisser le système s’équilibrer jusqu’à ce que la conductivité se stabilise ; la valeur affichée sera celle de l’EdM diluée 175x22 !
4. Commencer la titration en ouvrant le robinet de la burette contenant AgNO3 5.10-2 mol.L-1.
5. La conductivité décroît car AgCl précipite : les ions Cl– sont éliminés (ainsi que les ions Ag+) et sont remplacés par les ions nitrate (NO3–). Comme la conductivité molaire ionique de cet ion (7,142 mS.m2.mol-1) est inférieure à celle de l’ion chlorure (7,631 mS.m2.mol-1), le courant passe moins bien entre les électrodes de la cellule.
6. Lorsque le point d’équivalence est atteint (nAg+ = nCl–), la conductivité croît rapidement car AgNO3 n’est plus consommé.
7. Le graphe conductivité σ = f (VAg+ ajoutés) présente deux droites de pentes différentes. L’équivalence est repérée grâce au changement de pente des 2 droites. Leur intersection donne le volume équivalent VE (voir figure ci-dessous). Sur cette figure VE = 12,01 mL.

Calcul de la salinité :
A l’équivalence, la quantité d’halogénures totaux exprimés en mol.L-1 de Cl– présente initialement dans l’échantillon d’EdM mise dans le bécher et la quantité d’ions argent (Ag+) ajoutés sont dans les proportions stoechiométriques.
Soit :
CAg+ la concentration en AgNO3 de la solution titrante = 5.10-2 mol.L-1
VE le volume équivalent (en mL)
Vw le volume d’échantillon d’eau de mer
Cw la concentration en halogénures dans l’échantillon exprimée en mol.L-1 de Cl–
On a donc : CAg+ × VE = Vw × Cw, d’où
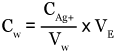
Or nous devons exprimer la chlorinité en g.kg-1. Comme la masse atomique du chlore est de 35,45 g.mole-1, la concentration devient : (Cw × 35,45) g.L-1.
La masse volumique (ρ) de l’échantillon d’EdM à 20°C étant mesurée préalablement, la chlorinité est donc :
(Cw × 35,45 × ρ-1) g.kg-1
La salinité étant égale à la chlorinité × 1,80655, le résultat final est de :
S = Cw × 35,45 × ρ-1 × 1,80655 g.kg-1.
8. Densimétrie/Réfractométrie/Conductimétrie
Rappel des notions correspondantes : voir Annexe 3.
Notes et Références
- Fabriquée à partir d’une solution à 20% (massique) du commerce. La masse volumique de cette solution étant de 1,098 kg.L-1, il est aisé (problème avec des élèves!) de trouver la molarité en HCl: 6,02M. Il faut donc diluer cette solution environ 300 x (3,3 mL dans 1L).
- Advanced Chemistry Sensor PASCO (https://www.pasco.com) , spécifiquement conçu pour une utilisation éducative.
- https://www.pasco.com/downloads/sparkvue
- https://www.pasco.com
- Il est donc recommandé de posséder des notions de base en informatique.
- La méthode complète est détaillée dans le manuel d’utilisation.
- La courbe dérivée apparait souvent irrégulière et les fluctuations apparaissant en dehors de la zone du point d’inflexion peuvent être négligées. Un lissage par moyenne mobile permet d’atténuer les irrégularités (Excel) et d’améliorer ainsi l’aspect du graphique.
- En chimie, un complexe est un édifice polyatomique constitué d’une ou de plusieurs entités indépendantes (ions ou molécules), en interaction. Ces complexes sont nommés « composés de coordination » et la chimie de coordination est la discipline qui les étudie.
- Solution du commerce 0,1 mol.L-1 diluée 10x.
- Une solution du commerce de NH3 à 2% est à 1,18 mol.L-1. Pour obtenir une solution équimolaire en NH4Cl, il faut dissoudre 63,13 g de ce composé dans un litre d’eau (1,18 mol.L-1).
- Il existe aussi des appareils appelés turbidimètres.
- Le titrage conductimétrique des ions sulfate dans l’eau minérale de Contrexéville: pas si simple ! S. Lampert. Le Bup n• 892.
- https://www.pasco.com/products/lab-apparatus/light-and-optics/ps-2600
- https://www.pasco.com/downloads/spectrometry
- Inspiré de http://chimactiv.agroparistech.fr/fr/aliments/analyse-eau/theorie-illustree/79
- Voir un cours de Chimie Organique.
- La quantité de chlore peut varier de quelques fractions de % selon les auteurs, mais le facteur de 1,80655 est généralement appliqué.
- Karl Friedrich Mohr, chimiste allemand, 1806-1879.
- Il s’agit d’une réaction photochimique provoquant la précipitation de l’argent réduit Ag0(s). Ce phénomène est à la base de l’ancienne industrie photographique. Il semble que le processus théorique de cette réaction soit encore mal connu.
- L’ajout d’eau distillée ne modifie pas la quantité d’ions Cl– à doser.
- Le nitrate d’argent est disponible auprès de fournisseurs de produits chimiques à usage scolaire, en droguerie ou en pharmacie. Masse molaire AgNO3 : 169,87 g.mol-1 ; 0,05 mol.L-1 = 8,5 g.L-1.
- La multiplication par le facteur de dilution de la valeur affichée au conductimètre donnera une valeur erronée de la conductivité réelle de l’échantillon car la linéarité de loi de Kohlrausch n’est plus vérifiée.
